This webpage accompanies the NIAB Diverse MAGIC wheat population, a mapping population of over 500 inbred lines with more than 1M SNPs imputed from whole genome sequence data and more than 70 phenotypes.
For a detailed description of our analysis, see
Scott MF, Fradgley N, et al. Limited haplotype diversity underlies polygenic trait architecture across 70 years of wheat breeding. Genome Biology 22, 137 (2021) https://doi.org/10.1186/s13059-021-02354-7.
The combination of carefully designed germplasm with dense phenotypic and genotypic data makes this a powerful and general purpose tool for dissecting trait genetic architecture.
For more information on multi-parent populations see Scott, Ladejobi et al. (2020).
Two webinars are available, an IWGSC webinar and a #UKPlantSciencePresents webinar where we discuss power and mapping multi-parent populations and then give an overview of the NIAB Diverse MAGIC wheat population.
Population Overview
The NIAB Diverse MAGIC population of winter wheat is derived from sixteen founders, which were chosen to maximise the genetic diversity captured from UK wheat varieties.
Variety
Year of Listing
Variety
Year of Listing
Variety
Year of Listing
Variety
Year of Listing
Holdfast
1935
Flamingo
1960
Stetson
1983
Soissons
1995
Steadfast
1942
Kloka
1965
Slejpner
1986
Robigus
2003
Bersee
1951
Maris Fundin
1975
Brigadier
1993
Cordiale
2004
Banco
1956
Copain
1980
Spark
1993
Gladiator
2004
MAGIC is an acronym for Multi-parental Advanced Generation Inter Cross.
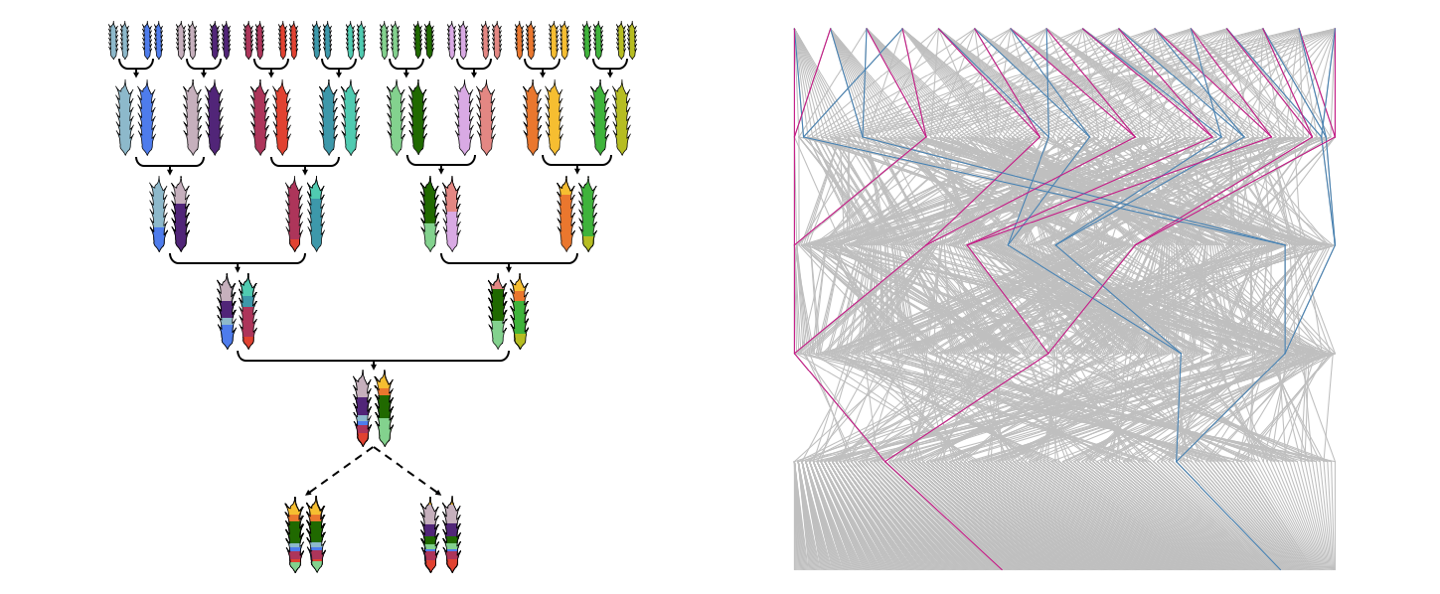
The sixteen founders were intercrossed in a funnel scheme and then selfed to produce inbred lines, called MAGIC lines.
A funnel crossing scheme is pictured here.
Each MAGIC line is derived from all the founders but receives a different mixture of genetic material.
Compared to biparental mapping populations, MAGIC populations capture greater genetic and phenotypic diversity by including multiple founders.
In addition, more recombination events occur over multiple rounds of inter-crossing, which allows finer mapping while minimising population structure.
The pedigree of the MAGIC lines in the diverse MAGIC wheat population is also shown here.
The genealogy of two different MAGIC lines is highlighted, demonstrating that the founders can be crossed in different combinations
Sequence, Genotype, and Phenotype Data
15 of the founders were sequenced after enriching for exome and promoter regions (~23x coverage of targets), one founder was sequenced using whole genome sequencing (~16x coverage of targets).
504 inbred lines were sequencing using ~0.3x whole genome sequencing. The sequencing data is available from the European Nucleotide Archive under project number PRJEB39021.
VCFs of the called SNP variation are available from the European Variant Archive (EVA).
SNP variants in exome/promoter regions were called from the founder data (EVA accession ERZ1643321).
At these SNP sites, we then made genotype calls in the inbred lines (i) by using the alignments to make genotype calls directly (EVA accession ERZ1643320) and (ii) by imputation with STITCH software (EVA accession ERZ1643322).
We provide files in PLINK format to give the genotypes of the founders and imputed genotypes of the inbred lines at ~1.1M sites. You can also download the inbred line genotypes at a tagging set of ~55k sites (LD pruned).
Imputation using the founder genotypes as the reference panel means that the founder that contributed genetic material at each locus in each inbred line is estimated.
There is some uncertainty in the reconstruction of these founder recombination mosaics, which can be accounted for by inferring the continuous 'dosage' of each founder at each locus.
We provide the inferred founder haplotype contributions at all 1.1M SNP sites for download here.
There is one .hap.txt file per chromosome giving the dosage at each site for each founder (column K, numbered in alphabetical order) for each inbred line.
These were extracted from the VCFs outputted by STITCH using this script.
Phenotypes for the founders and inbred lines are available to download here. Phenotype descriptions and collection methods are described in Scott, Fradgley et al. (2020).
The diverse MAGIC wheat population has also been characterised using the 35k Wheat Breeders' Array.
Genotyping calls are available to download for the 16 founders and for 550 MAGIC lines.
Calls were made using the Axiom Best Practices Genotyping Analysis workflow with an inbreeding penalty of 4. The released genotypes have consensus calls where multiple samples were genotyped from the same line.
In addition, the genotypes at sites with no minor homozygous calls have been adjusted.
These genotype and phenotype data are also available from the UCL data repository .
Scripts for alignment, variant calling, imputation, and association mapping are provided in the GitHub Repository michaelfscott/DIVERSE_MAGIC_WHEAT.
A genetic map for the trimmed set of 55k SNPs is available here. This genetic map was constructed using the 'qtl2' R package (Broman et al. 2019) as described in Fradgley et al., 2022
Analysis
We provide a simple pipeline for genetic mapping with these data.
Download this directory. The 'DATA' subdirectory contains the phenotypic data and the tagging set of ~55k SNP sites called in 504 inbred lines.
In this directory, we include R functions for association mapping (file mixed.model.functions.r), including a mixed model transformation to remove the inflationary effects of unequal relatedness on genetic associations. Association mapping can be run on the basis of SNPs or the inferred founder haplotype dosages.
To run, follow the steps in the R script example.analysis.r (this will run without modification if the downloaded directory is used as the R working directory).
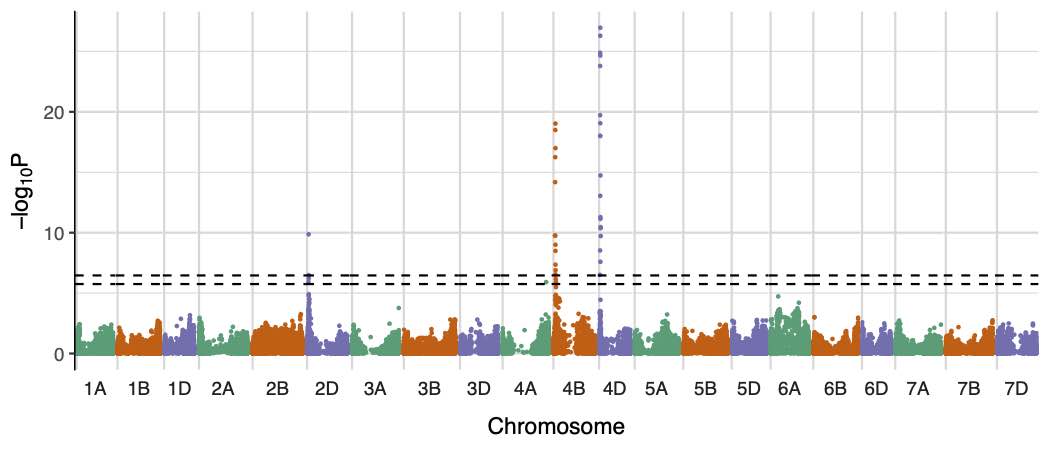
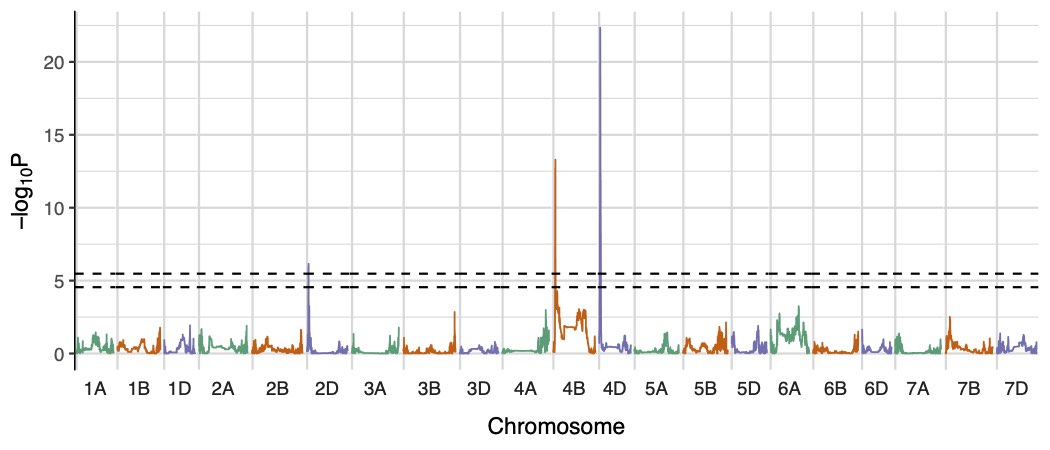
We also include a function for plotting the results as a manhattan plot (plot.functions.r). Example output manhattan plots for 'height to ear tip' are shown here.
Manhattan plots for SNP-based and founder-haplotype-based association tests on the 'height to ear tip in year 2' phenotype.
New phenotypes can be analysed by creating a new tab-separated file with the sample names in one column and the phenotype in another column.
The name of the column names is assumed to be 'line_name', but this can be adjusted by setting the 'id.column' variable.
The name of the column containing the phenotype should be specified using the 'phenotype' variable.
About
The diverse MAGIC wheat population was developed at the National Institute for Applied Botany (NIAB), from whom germplasm is available (contact James Cockram).
Genetic resources for this population were developed in collaboration between University College London (UCL) and NIAB as part of the MAGIC CARPeT project (MAGIC: Community Access to Resources, Protocols and Training), funded by the Biotechnology and Biological Sciences Research Council (BBSRC) under grants BB/M011666/1 (to NIAB) and BB/M011585/1 (to UCL).
 Close
Close